10강. 게놈 프로젝트와 시퀀싱 기술
추천글 : 【생물학】 생물학 목차
a. 파이로시퀀싱
b. 후성유전체 시퀀싱
c. 생물정보학 분석 목차
d. 전사체 분석 파이프라인
1. 게놈 프로젝트(genome project) [목차]
⑴ 개요
① 1990년 왓슨 및 Francis Collins가 지휘하면서 시작 : 6개국 연합의 15년 과제로 출범
② 350여 연구기관 공동 연구
○ 2000년 6월 11일 84.5 % 완성, 초안 발표
○ 2003년 4월 15일 99.99% 정확도의 완결편 발표
○ 2,800명 이상의 연구자가 13년간 참여하였으며 $2.7 billion이 소요됨
③ 인간유전체 연구의 부수효과
○ 생물정보학의 탄생
○ 인체 단백질 생산 공정의 개발을 촉진
○ 인슐린(insulin) : 최초로 서열이 결정된 단백질
○ 자동화된 염기서열 분석기기 개발을 촉진
○ 응용성 있는 다른 생물의 유전체 분석 촉진
⑵ 방법론 1. 단계적 서열분석법 : 과학자 진영
① 단계 1. 제한효소 인식 부위 결정
○ 제한효소로 DNA를 자른 뒤 전기영동을 하면 각 절편의 크기를 알 수 있음
○ 두 개의 제한효소를 여러 방식으로 처리하면 두 효소의 제한효소 인식부위의 상대적 거리를 알 수 있음
② 단계 2. 유전자 지도 작성
○ 염색체 상의 유전자의 상대적 거리를 결정하는 것
○ 교차율을 통해 유전자 사이의 거리를 추론할 수 있음
③ 단계 3. 물리적 지도 (DNA 지도) 작성
○ 제한효소 인식 부위 결정의 의미 : 각 제한효소 인식 부위를 지도 삼아서 염기순서를 알아낸 단편들의 정보로 물리적 지도를 누적적으로 작성
○ 유전자 지도 작성의 의미 : 물리적 지도가 작성되면 유전자 지도와 대조할 수 있음. 유전자 사이에는 인트론이 있음
○ 하나의 라이브러리로 접근하는 방식
⑶ 방법론 2. 산탄식 서열분석법 (Celera, J. Craig Venter) : 기업가 진영
① 하나의 DNA를 여러 방법으로 자름
② 하나의 방법으로 잘린 토막들의 염기 순서를 모두 밝혀냄
○ 분석 시료의 길이는 제한돼 있으므로 한 번에 DNA 순서를 알 수는 없음
③ 각각의 방법에서 염기들의 배열을 무작위적으로 하여 공통된 결과를 이를 때까지 계속
④ 컴퓨터 사이언스에 기반한 방법
⑷ 과학자 진영 vs 기업가 진영
① 과학자 진영 : The Human Genome Project (HGP). 왓슨과 Francis Collins. 최종 비용은 $2.7 billion
② 기업가 진영 : Celera Genomics (1998년 시작). Craig Venter. 최종 비용은 $300 million
③ 두 진영 모두 인간 게놈의 초안을 2001년에 공개함
④ 과학자 진영은 기업가 진영이 자신들의 공로와 투자를 가로채 가는 것이 불만
⑤ 기업가 진영은 과학자 진영이 정보를 공개하지 않는 것이 불만 → 새로운 방법론 개발
⑥ 게놈 지도의 최종 완성은 둘 모두의 공으로 합의

Figure. 1. 단계적 서열 분석법과 산탄식 서열 분석법
2. 시퀀싱 기술 [목차]
⑴ 개요
① DNA 시퀀싱 : DNA 복제 원리를 응용
○ 주형 : DNA의 각 사슬
○ 기질 : dNTP(dATP, dCTP, dGTP, dTTP)
○ NTP는 2' 탄소에 -OH기가 달려있는 것으로 RNA 중합의 재료
○ DNA 중합효소 : 디옥시리보오스의 3‘-OH에 다음 뉴클레오티드의 인산이 결합
○ 합성 방향 : 5‘ → 3’, 주형과 상보적인 염기쌍 형성
○ ddNTP : 3번 탄소가 OH기가 아니라 H기이므로 DNA 중합이 중단
② RNA 시퀀싱 : RNA 전사 원리를 응용
③ sequencing by synthesis
○ 정의 : 중합효소가 프라이머 서열에 뉴클레오타이드를 첨가할 때 신호를 발생시켜 염기서열을 결정하는 방식
○ 종류 1. CRT(cyclic reversible termination) : incorporation, imaging, deprotection이 사이클을 이루는 방식
○ 종류 2. SNA(single nucleotide addition) : 한 번에 한 개의 뉴클레오티드만 추가됨
○ 예 : ion semiconductor sequencing (PGM), pyrosequencing (Roche 454)
④ sequencing by ligation
○ 정의 : 리게이즈가 hybridized sequence를 primer sequence에 연결할 때 신호를 발생시켜 염기서열을 결정하는 방식
○ 한 번에 1개 이상의 뉴클레오티드를 첨가할 수 있음 (e.g., dibase)
○ 예 : nanoball sequencing, Thermo Fisher SOLiD
⑵ in vitro cloning : 가장 최초의 시퀀싱 방법
⑶ 디데옥시 사슬 종결법 (= Sanger sequencing) : 1977년 보고, 생어(Sanger)의 두 번째 노벨상
① 기질 : dNTP + ddNTP 소량 + buffer (pH 안정)
○ ddNTP는 3'-OH기가 없으므로 중합 반응을 종료시킴
○ 만약 ddNTP를 많이 넣으면 모든 주형 DNA가 단시간에 중합을 중단
② 프라이머
○ 예 : p32-프라이머 (CTAG)
③ 1st. 주형 DNA와 중합효소 첨가
④ 2nd. 중합반응 후 가열하여 복제 가닥 분리
⑤ 3rd. 전기영동 후 X선 필름에서 서열 판독 하거나 형광 조사
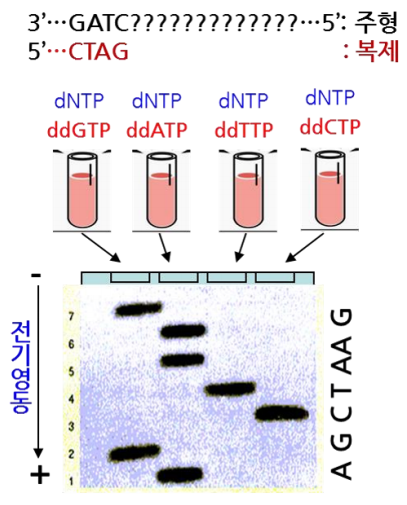
⑥ 장점 : 아주 긴 가닥도 읽을 수 있어 아직도 실험실에서 활용됨
⑦ 단점 : 같은 DNA 가닥이 매우 많아야 함
⑷ dye-디데옥시 사슬 종결법 : 레이저 이용
① dNTP에 4색 형광 - ddNTP 소량 첨가
② 자동 염기서열 판독 가능
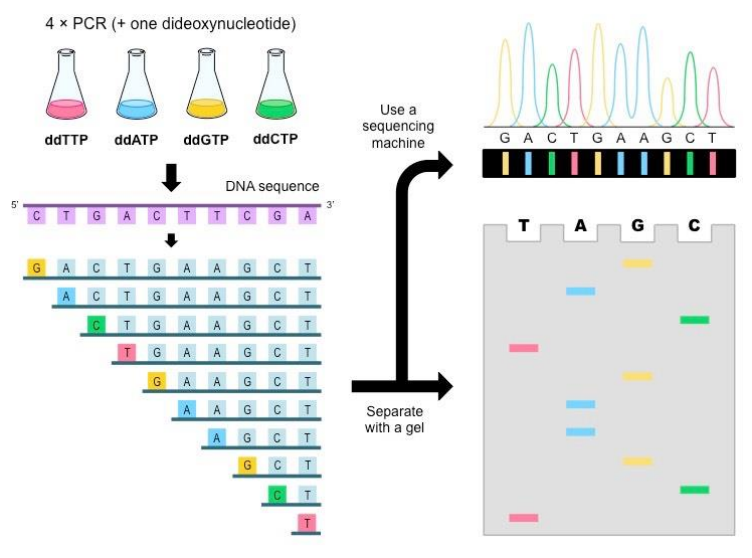
Figure. 3. Dye-디데옥시 사슬 종결법의 과정
⑸ ion semiconductor sequencing (PGM)
① sequencing by synthesis
② insertion / deletion 에러가 많음
⑹ 파이로시퀀싱(pyrosequencing, Roche 454)
① 정의 : DNA 중합 시 나오는 피로인산의 양에 비례하여 발광하도록 함으로써 수행되는 DNA 서열결정법 (~400 bp / read)
② sequencing by synthesis : 뉴클레오티드가 결합하면 빛이 발생하고 pyrogram에서 피크로 나타남
③ insertion / deletion 에러가 많음
④ 더이상 사용되지 않음
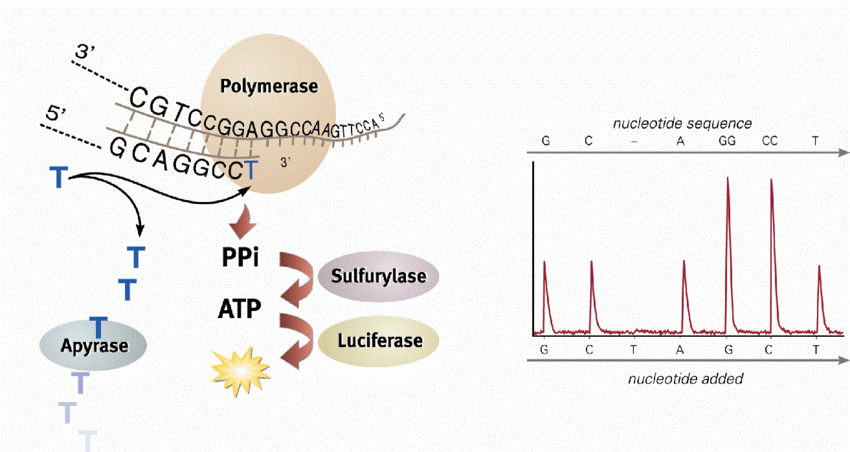
Figure. 4. 파이로시퀀싱 모식도
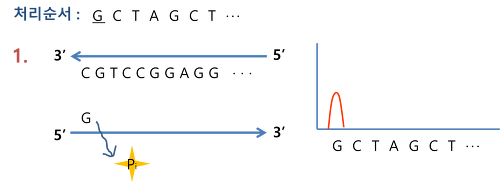
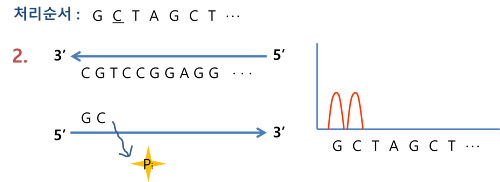
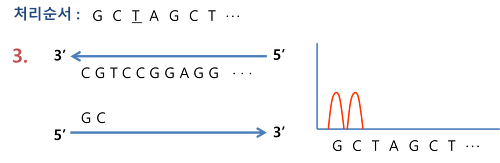


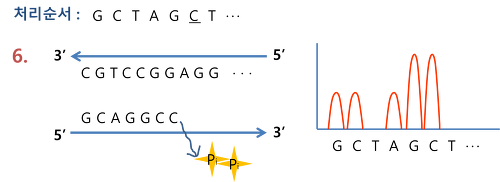
Figure. 5. 파이로시퀀싱 과정
⑺ nanoball sequencing
① sequenching by ligation
⑻ Thermo Fisher SOLiD
① sequenching by ligation
② 더이상 사용되지 않음
⑼ Illumina solid-phase amplication (ref, ►)
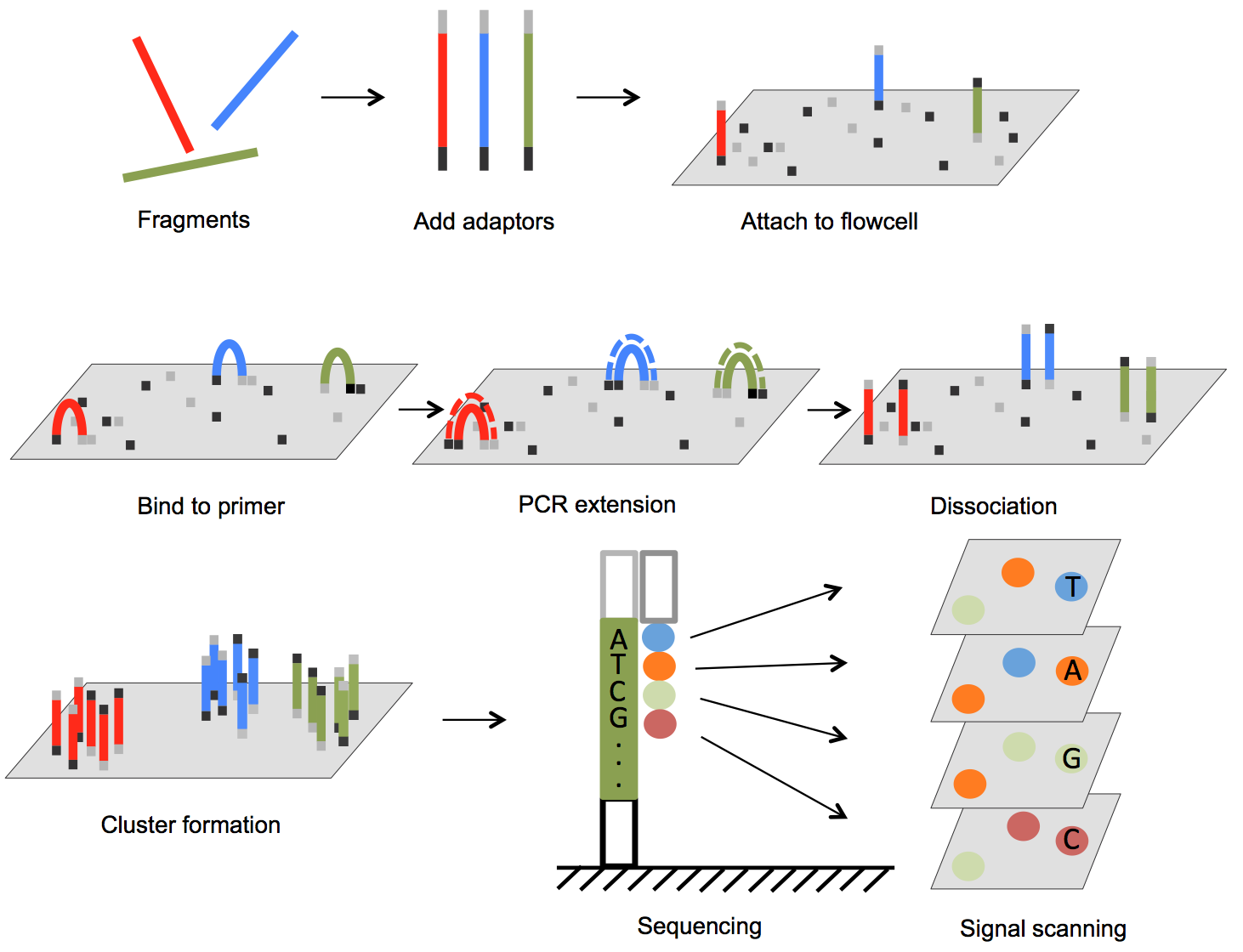
Figure. 6. Illumina solid-phase amplication

① 1st. 절편화 : 주어진 DNA 시료를 랜덤하게 자름
② 2nd. gel-based size selection : 필요한 경우 각 DNA fragment의 사이즈를 한정할 수 있음
③ 3rd. adaptor binding : 모든 DNA 시료 절편의 양 말단에 어댑터를 붙임. transposome을 이용
④ 4th. amplification
○ 4th - 1st. DNA를 single-strand로 변성
○ 4th - 2nd. single-stranded DNA를 illumina flow cell에 부착
○ 4th - 3rd. 효소를 첨가하여 solid-phase substrate 상에서 single-stranded DNA가 bridge가 되도록 함
○ 4th - 4th. single-stranded DNA bridge에 프라이머를 첨가하면 프라이머가 그 bridge에 결합할 수 있음
○ 4th - 5th. unlabeld single-stranded DNA를 첨가한 뒤 DNA 중합반응 유발 : double-stranded DNA bridge가 됨
○ 4th - 6th. denaturing을 통해 double-stranded DNA bridge가 anchored single-stranded DNA가 되도록 함
○ 4th - 7th. 위 6개의 과정을 반복하여 동일한 염기서열을 가지는 anchored single-stranded cluster를 생성
○ 특징 : anchored single-stranded cluster는 몇백만 cluster를 형성함
⑤ 5th. unwanted priming을 방지하기 위해 forward strand만 남김
⑥ 6th. sequencing by synthesis (SBS)
○ 6th - 1st. 염기 종류에 따른 4종류의 labled reversable terminator, 프라이머, DNA 중합효소를 첨가
○ 6th - 2nd. labled reversable terminator 뉴클레오티드가 이인산에스테르 결합을 형성하면 형광이 발생
○ 6th - 3rd. 각 클러스터의 형광 색깔 분포를 사진으로 획득
○ 6th - 4th. washing
○ 6th - 5th. 위 4개의 과정을 반복하여 전체 염기서열을 결정
○ 종류 1. single-end 시퀀싱 (SES) : 어댑터의 한쪽만으로 시퀀싱을 하는 방식
○ 종류 2. paired-end 시퀀싱 (PES) : 어댑터의 양쪽으로 시퀀싱을 하는 방식
○ 우선 한 어댑터를 통해 시퀀싱을 하고 (Read1 획득), 그 뒤 반대쪽 어댑터를 통해 시퀀싱을 함 (Read2 획득)
○ 한 DNA fragment에서 나오는 Read1과 Read2는 동일한 클러스터에서 나오므로 쉽게 둘을 대응시킬 수 있음
○ 장점 : 더 높은 정확성 (∵ Read1, Read2 상호 간 비교), DNA 변이 도출 용이, 반복서열 분석 용이, 이종 간 맵핑 용이
○ 단점 : 더 비싼 가격, SES보다 스텝이 더 많이 필요함
⑦ ~100 bp / read : 최근에는 250 bp
⑽ WGS(whole genome sequencing)
① SNV, insertion, deletion, structural variant, CNV
② sequencing depth > 30X
⑾ WES(whole exon sequencing)
① 오직 protein-coding gene에 대한 SNV, insertion, deletion, SNP
② sequencing depth > 50X ~ 100X
③ 비용이 저렴함
⑿ RNA-seq
① 개요
○ 마이크로어레이 데이터는 raw 데이터가 연속형 데이터인데 RNA-seq은 raw 데이터가 count data
○ RNA-seq은 마이크로어레이 데이터에 비해 발현이 너무 낮거나 높은 유전자에 대해서도 robust하게 신호를 얻을 수 있음
② 1st. microdissection : RNA 추출을 위해 특정 조직을 분리하는 것
○ LCM(laser capture microdissection) : 레이저 빔으로 특정 조직을 절단. robust하지만 labor intensive하다는 단점
○ TOMO-seq : cryosection을 이용하고 컴퓨터를 통해 3D sectioning을 할 수 있음. 임상적 목적으로는 사용할 수 없음
○ transcriptome in vivo analysis
○ ProximID
○ STRP-seq
③ 2nd. RNA의 poly A 꼬리를 인식하는 poly T를 결합시킴
④ 3rd. RNA를 절편화 : 200-400 nt
⑤ 4th. RNA에 프라이머 부착
⑥ 5th. 첫 번째 cDNA 합성
⑦ 6th. 두 번째 cDNA 합성
⑧ 7th. RNA의 3'과 5' 말단에 가공
⑨ 8th. DNA sequencing adapters ligated
⑩ 9th. ligated 절편을 PCR로 증폭
⑪ 응용 1. dUTP method : strand-specific sequencing의 대표적인 방법
○ 배경 : RNA 방향성에 따른 생물학적 기능을 연구할 때 사용 (예 : antisense miRNA의 조절)
○ step 1. DNA&RNA hybrid : mRNA poly-A tail에 붙는 dT 프라이머와 역전사 효소로 cDNA (first or anti-sense strand) 합성
5'-//-U-//-AAAAAA-3'
3'-//-A-//-TTTTTT-5'
○ step 2. ds cDNA : dTTP 대신 dUTP를 이용하여 cDNA (first strand)를 주형으로 cDNA (second or sense strand)를 합성
3'-//-A-//-TTTTTT-5'
5'-//-U-//-AAAAAA-3'
○ step 3. ligated ds cDNA : Y-adaptor를 ds cDNA 양쪽에 연결
○ step 4. UDG(uracil-DNA glycosylase)를 처리하면 우라실을 포함하는 DNA인 second strand가 분해됨
○ step 5. 남아 있는 reverse antisense strand인 first strand를 증폭해 라이브러리를 생성
○ 라이브러리 raw 데이터에서 "_1.fastq"가 first strand이고, "_2.fastq"가 second strand
○ 즉, _2.fastq가 원래 RNA의 프로파일을 나타냄
⒀ 단일세포시퀀싱(single cell sequencing)
① 종류
○ scDNA-seq
○ scRNA-seq (2013년 올해의 기술) : Chromium, Smart-seq 등
○ single cell epigenetics sequencing
② 단계 1. 단일 세포 분리
○ 방식 1. 단순한 분리 : 매우 초창기 방식
○ 방식 2. FACS 또는 LCM(laser microdissection) 기반
○ 방식 3. acoustic separation
○ 유체역학적으로 단일 세포를 구분하므로 세포에 영향을 비교적 적게 줌
○ 대표적으로 CyTOF(cytometry by time of flight)가 있음
○ 방식 4. immuno-magnetic separation
○ cell에 magnet을 붙임
○ 세포를 많이 획득할 수 있음
○ centrifugation이 필요한 경우와 그렇지 않은 경우로 구분
○ 크게 droplet-based platform과 plate-based platform으로 나눌 수 있음
③ 단계 2. reverse transcription
④ 단계 3. cDNA amplification
⑤ 단계 4. 라이브러리 구성 : Drop-seq 등
⑥ 단일세포유전체(scDNA-seq) + 단일세포전사체(scRNA-seq)
○ 유전체 상의 mutation pattern이 전사체 상의 gene expression과 어떻게 연관되는지 알 수 있음
○ DNA와 RNA를 분리하는 기술 : G&T seq, SIDR-seq, DNTR-seq
⒁ 단일핵전사체(single nucleus RNA sequencing, snRNA-seq)
① 목적 1. 근육은 다핵세포이므로 scRNA-seq에 의해 캡처되지 않으므로 세포보다는 핵 단위로 분석할 필요가 대두됨
② 목적 2. scRNA-seq보다 intron, pre-mRNA, non-coding RNA 등 다양한 RNA가 많이 잡힘
③ 목적 3. snRNA-seq에서 핵 RNA가 주로 많이 잡힘 : 세포질 RNA가 (소량이지만) 잡히긴 함
④ 목적 4. snRNA-seq은 대부분 nascent RNA이므로 scRNA-seq보다 세포의 상태를 더 잘 반영함
⒂ 공간적 시퀀싱(spatial resolved sequencing) (▶ 추가 설명)
① 개요
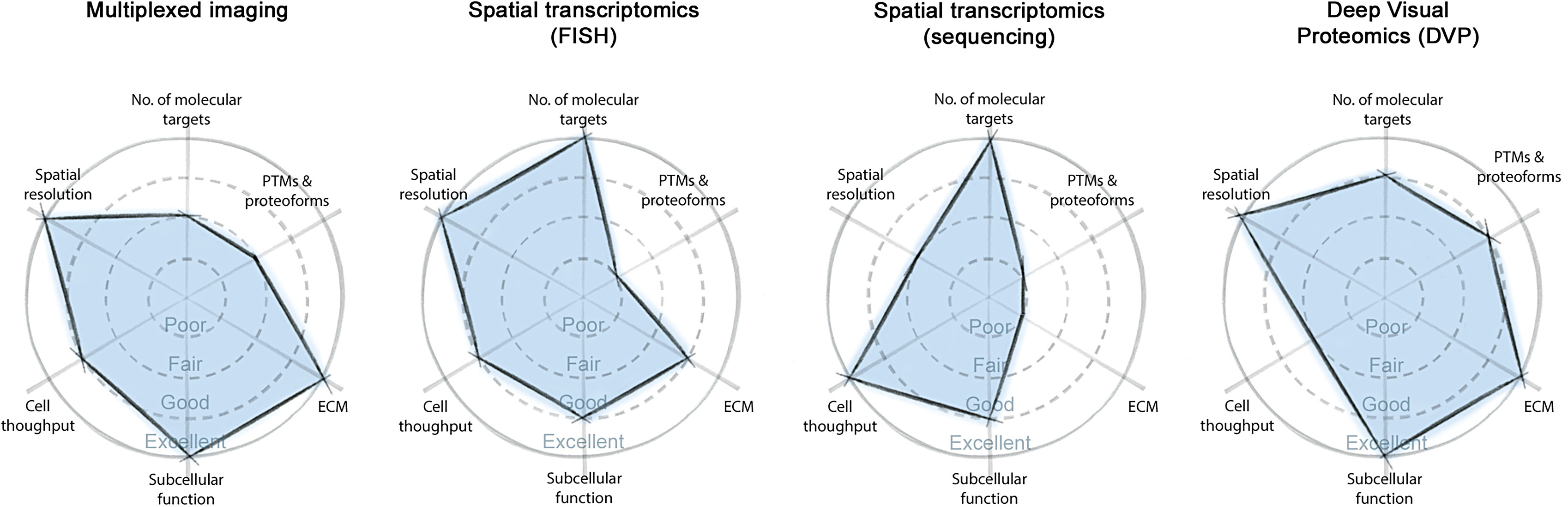
Figure. 8. 공간적 시퀀싱 개요
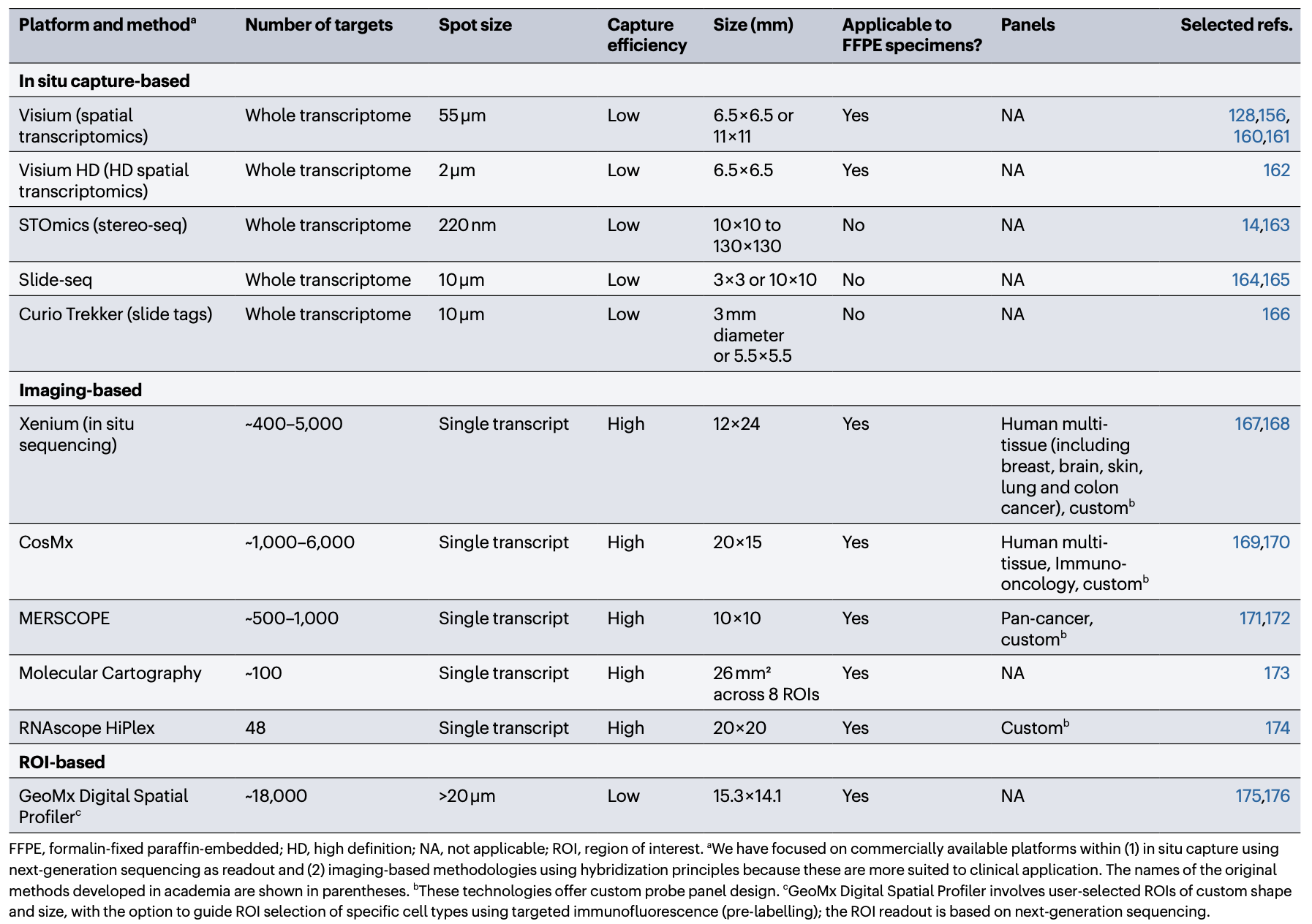
Table. 1. 공간적 시퀀싱 비교
| 연도 | 방법 | 샘플 | 타겟 | 해상도 | 단일 세포 | 프로브 | 면적 | 세포수 | 유전자수 |
| 스팟 기반 공간전사체 | |||||||||
| 2014 | Tomo-seq | fresh-frozen | transcriptome-wide | - | no | - | - | - | 12,000 genes per section |
| 2016 | Visium | fresh-frozen and FFPE | A-tailed RNA transcripts and targeted genes | 100 μm | no | 6.5 mm × 6.5 mm | 20,000 ~ 40,000 cells | ~1.700 genes per spot | |
| 2017 | Geo-seq | fresh-frozen | transcriptome-wide | - | yes | - | - | 5-40 cells per section | ~8,000 genes |
| 2019 | Slide-seq/V2 | fresh-frozen | A-tailed RNA transcripts | 10 μm | no | beads | Φ3.0 mm | - | 550 UMI per read |
| 2019 | HDST | fresh-frozen | A-tailed RNA transcripts | 2 μm | no | beads | 5.7 mm × 2.4 mm | ~60,000 cells per individual hexagonal | 44 UMI per 5× beads |
| 2020 | DBiT-seq | FFPE | A-tailed RNA transcripts | 25 μm | no | free probe | 2.5 mm × 2.5 mm | ~50,000 cells | 1,000 genes per pixel |
| 2021 | Seq-Scope | fresh-frozen | A-tailed RNA transcripts | ~0.6 μm | yes | in situ synthesize | 0.8 mm × 1 mm | - | ~1,617 genes per cell |
| 2021 | PIC | fresh-frozen | transcriptome-wide | 75-5,000 μm | yes | photo-caged oligonucleotides | scales with acquisition time | 10 or more cells in tissue sections | ~8,000 genes |
| 2021 | sci-Space | fresh-frozen | A-tailed RNA transcripts | 222 μm | yes | beads | 18 mm × 18 mm | 121,909 cells | 1,231 genes per cell |
| 2022 | Stereo-seq | fresh-frozen | A-tailed RNA transcripts | 0.5 μm | yes | in situ synthesize | up to 132 mm × 132 mm | 100,000-16,900,000 cells | 1,910 UMI and 792 genes per cell |
| 2022 | Pixel-seq | fresh-frozen | A-tailed RNA transcripts | ~1 μm | yes | in situ synthesize | 75 mm × 25 mm | 15,000 cells per section | ~500 genes per cell |
| 2023 | Slide-tags | fresh-frozen | A-tailed RNA transcripts | 10 μm | yes | beads | Φ3.0 mm | 81,000 cells | 2,377 UMI per cell |
| 2024 | Visium HD | fresh-frozen, FFPE | targeted transcriptomics | 2 μm | yes | 6.5 mm × 6.5 mm | 20,000-40,000 cells | 7,605 probes | |
| 2024 | Open-ST | fresh-frozen | A-tailed RNA | 0.6 μm | yes | in situ synthesize | 6.3 mm × 89 mm | - | ~1,000 UMI per cell |
| 이미지 기반 공간전사체 | |||||||||
| 2013 | ISS | cells, tissue sections | targeted RNA | - | yes | padlock probe | 222 × 166 μm² | 450 cells covering an area of 0.16 mm² | 39 genes |
| 2014 | FISSEQ | cells, tissue sections | untargeted RNA | - | yes | fluorescent probe | 10 mm × 10 mm | - | 4,171 genes with size >5 pixels |
| 2015 | MERFISH | cells, tissue sections | targeted RNA | - | yes | oligonucleotide probes | scales with acquisition time | ~100, up to 40,000 cells | 130 genes, up to 10,050 genes |
| 2018 | STARmap | tissue sections | targeted RNA | - | yes | padlock probe | 1.7 mm × 1.4 mm × 0.1 mm | > 30,000 cells | 160 to 1,020 genes per sample |
| 2019 | seqFISH+ | cells, tissue sections | targeted RNA | - | yes | oligonucleotide probes | scales with acquisition time | 2,963 cells | 10,000 genes |
| 2021 | ExSeq | cells, tissue sections | untargeted / targeted RNA | - | yes | padlock probe | 0.933 mm × 1.140 mm × 0.02 mm | ~2,000 cells | - |
| 2021 | EASI-FISH | tissue sections | targeted RNA | 0.23 × 0.23 × 0.42 μm | yes | - | - | 80,000 cells | - |
| 2022 | EEL FISH | tissue sections | targeted RNA | - | yes | primary probe | scales with acquisition time | 128,000 cells | 883 genes |
| 2023 | STARmap PLUS | tissue sections | targeted RNA and antibody-based protein | - | yes | padlock probe | 194 mm × 194 mm × 345 mm | - | 1,022 genes |
| 멀티오믹스 기반 | |||||||||
| 2018 | CODEX | cells, tissue sections | antibody-based protein | - | yes | - | - | - | - |
| 2020 | OligoFISSEQ | cells, tissue sections | genomics | - | no | barcoded oligopaint probes | - | - | - |
| 2020 | DBiT-seq | fresh-frozen | A-tailed RNA transcripts and antibody-based protein | 10 / 25 / 50 μm | no | free probe | 2.5 mm × 2.5 mm | ~50,000 cells | ~4,000 genes per 50 μm pixel |
| 2021 | DNA seqFISH+ | cells | genomics | N/A (5,616.5 ± 1,551.4 dots per cell) | yes | primary probe | scales with acquisition time | 446 cells | 3,660 chromosomal loci per cell |
| 2021 | IGS | cells, tissue sections | genomics | 400-500 nm | yes | hairpin DNA | - | 113 cells | - |
| 2021 | slide-DNA-seq | fresh-frozen | genomics | 10 μm | yes | beads | Φ3 mm | 2,274 cells | - |
| 2022 | CAD-HCR | cells | targeted RNA and antibody-based protein | - | yes | padlock probe | - | - | - |
| 2022 | MOSAICA | cells, FFPE | targeted RNA and antibody-based protein | - | yes | primary code | - | - | - |
| 2022 | SM-Omics | fresh-frozen | A-tailed RNA transcripts and antibody-based protein | 55 μm | no | 6.5 mm × 6.5 mm | - | - | |
| 2022 | Spatial-CUT&Tag | fresh-frozen | epigenomics | 20 μm | no | - | - | - | H3K27me3: 9,735; H3K4me3: 3,686 per 20 μm pixel size |
| 2022 | Spatial-TREX | fresh-frozen | A-tailed RNA transcripts and CloneIDs | single cell/55 μm | yes | - | - | 65,160 cells | 5,000-10,000 genes |
| 2022 | Perturb-map | fresh-frozen | A-tailed RNA transcripts and CRISPR-targeted genes | 55 μm | yes | - | - | - | - |
| 2022 | slide-TCR-seq | fresh-frozen | A-tailed RNA transcripts and targeted T cell receptor genes | 10 μm | no | beads | Φ3.0 mm | - | - |
| 2022 | SmT | fresh-frozen | host transcriptome- and microbiome-wide | 55 μm | no | 6.5 mm × 6.5 mm | - | - | |
| 2022 | RIBOmap | cells, tissue sections | ribosome-bound mRNAs | - | yes | tri-probes (split DNA probe, padlock probes, primer probes) | - | 60,481 cells | 5,413 genes |
| 2022 | epigenomic MERFISH | cells, tissue sections | epigenomics | - | yes | oligonucleotide probes | scales with acquisition time | ~5,400 cells | 3 histone modifications, H3K4me3: 127 genes/loci; H3K27ac: 142 target genomic loci |
| 2023 | spatial ATAC | fresh-frozen | epigenomics | 55 μm | no | splint oligonucleotide, spatially barcoded surface oligonucleotides | - | - | - |
| 2023 | spatial-CITE-seq | fresh-frozen | A-tailed RNA transcripts and antibody-based protein | 50 μm | no | free | 2.5 mm × 2.5 mm | 37,500-50,000 cells | 411 genes and 153 proteins per pixel |
| 2023 | Stereo-CITE-seq | fresh-frozen | targeted RNA and antibody-based protein | 500 nm | yes | in situ synthesize | up to 132 mm × 132 mm | 100,000-16,900,000 cells | 4.05K UMI per bin50 |
| 2023 | Spatial-ATAC-RNA-seq | fresh-frozen | A-tailed RNA transcripts and transposase chromatin | 20 / 50 μm | no | free | 2.5 mm × 2.5 mm | 37,500-50,000 cells | - |
| 2023 | Ex-ST | fresh-frozen | A-tailed RNA transcripts | 20 μm | no | 6.5 mm × 6.5 mm | 8,000-16,000 cells | ~500 gens per spot | |
| 2023 | immuno-SABER | cells | antibody-based protein | - | yes | - | - | - | - |
| 2023 | scDVP | fresh-frozen | proteins | - | no | - | - | - | 1,700 proteins |
| 2023 | scSpaMet | fresh-frozen, FFPE | metabolomics and targeted multiplexed protein | - | yes | - | - | - | - |
Table. 2. 공간적 시퀀싱 비교
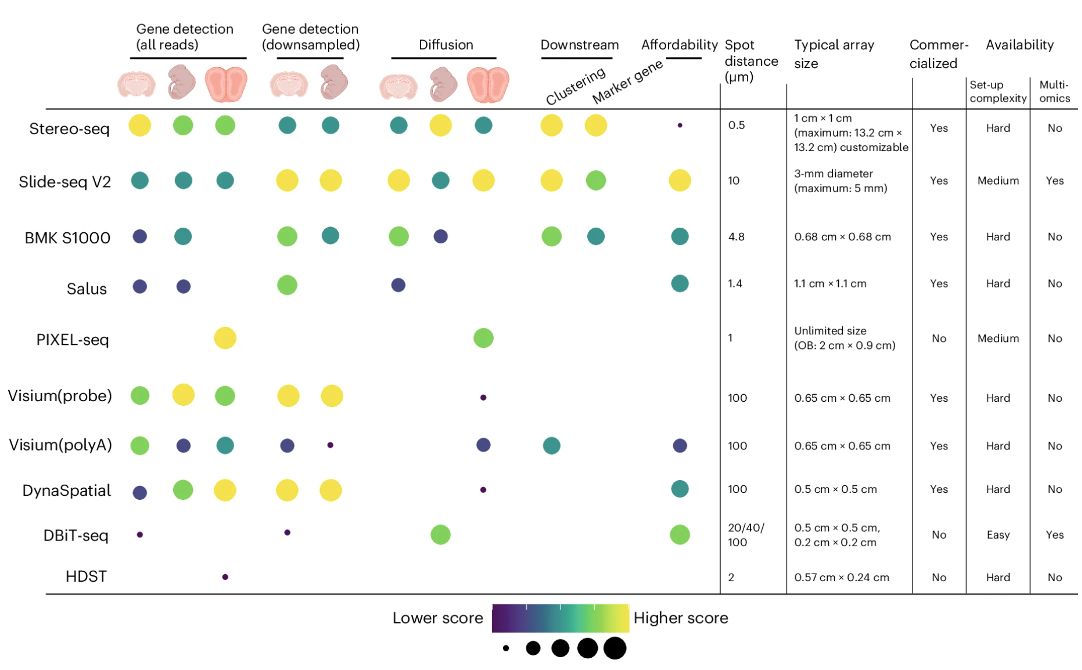
Figure. 9. 스팟 기반 공간전사체 간 비교
② 종류 1. 공간유전체(spatial genomics)
○ 예 1. 종양 조직 : 종양은 불균일성이 있으므로 (cf. CNV(copy number variation))
○ 예 2. 비장 연구 : 성숙한 면역세포는 유전자 구성이 모두 다르므로
③ 종류 2. 공간전사체(spatial transcriptomics) : 2020년 올해의 기술
○ 예 1. 대뇌 피질 : white matter, gray matter, L6, L5, L4, L3, L2, L1
○ 예 2. 비장 : red pulp, white pulp, intermediate zone
○ 예 3. 피부 : epidermis, dermis, smooth muscle
○ 예 4. 신장 : cortex, Henle loop, collecting duct
○ 예 5. 간 : periportal, periportal, intermediate
○ 예 6. 고환 : I-III, IV-VI, VII-VIII, IX-XII
○ 예 7. 나뭇잎 : upper epidermal wall, lower epidermal wall
④ 2-1. 스팟 기반 공간전사체 (spatial indexing transcriptomics) : 많은 유전자 + 적은 스팟
○ ST (spatial transcriptomics)
○ barcoded oligo를 랜덤하게 조직에 뿌린 뒤 각 조직으로부터의 mRNA를 capture 하는 방식
○ 10X Visium
○ 원리 : 각 스팟에 스팟 특이적인 oligonucleotide를 부착시켜 조직 유래 RNA와 혼성화 → spotwise transcriptome 획득
○ 표면적 : 6.5 mm × 6.5 mm
○ 두께 : 10 ~ 20 μm
○ 스팟의 개수 : 최대 4992개 (Visium HD 이전 버전 기준)
○ 스팟 간 거리 : 100 μm
○ 스팟의 직경 : 55 μm
○ sensitivity : 10,000 transcripts per spot
○ 종류 1. direct Visium (oligo-dT based method)
○ poly dT로 mRNA를 capture
○ FF(fresh-frozen) 샘플에만 적용 가능 : FFPE에 쓰이는 시약이 direct Visium에 적합하지 않음
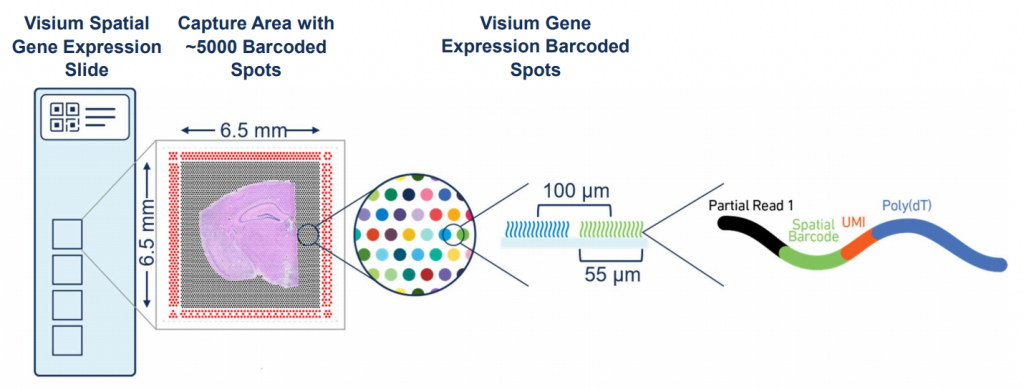
Figure. 10. Visium FF 원리
○ 종류 2. probe-based Visium
○ FF, FFPE(formalin-fixed paraffin-embedded) 모두에서 할 수 있음 : 특히 FFPE 샘플은 mRNA들이 여러 파편들로 끊어져 있는 등 RNA degradation이 있어서 direct Visium을 할 수 없어 중요함
○ 3쌍의 LHS와 RHS가 모두 붙어야 target mRNA가 식별됨 : 각 프로브의 길이는 25 bp. RTL(probe-based RNA-templated ligation chemistry)을 이용
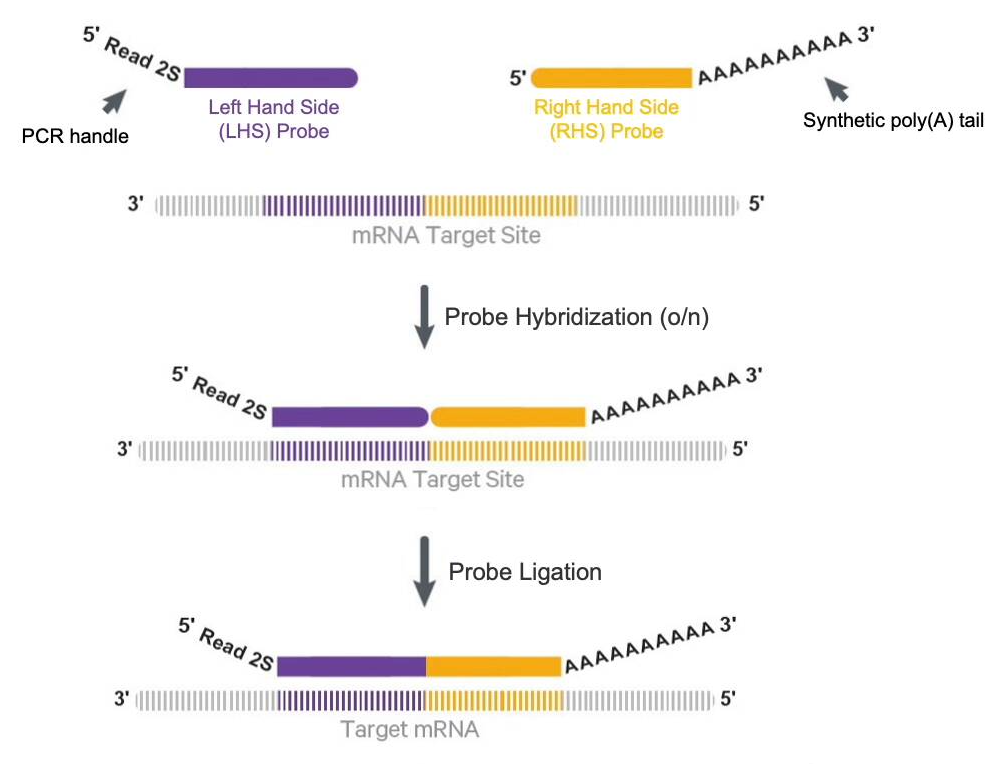
Figure. 11. probe-based Visium 원리
○ 장점 : direct Visium보다 데이터 퀄리티가 좋음
○ 단점 : probe 기반으로 지정된 유전자만 detect를 하므로 Visium FF보다 분석 상의 자유도가 떨어짐
○ 2024년 6월부터 10x는 CytAssist가 아닌 Visium FFPE 서비스를 종료
○ CytAssist 이미지는 gene expression 분포를 나타내어 image alignment에 쓰임
○ 10X Visium HD
○ 직경 2 μm 스팟 단위로 구성된 데이터가 기본 데이터이며 8 μm, 16 μm로 binning한 데이터도 추가로 제공됨
○ Slide-seq (slideSeq), Slide-seq V2 (직경 10 μm)
○ 랜덤하게 spatial bead를 뿌리고 in-situ sequencing을 하는 방식
○ 스팟의 97%가 하나 혹은 두 개의 셀 타입으로 구성돼 있음
○ 세포핵 자체를 labeling하므로 nuclear segmentation이 필요하지 않음
○ Curio Seeker : Slide-seq V2의 상용화 버전
○ HDST
○ 바코드가 새겨진 bead를 patterned wafer 위에 올리고 serial hybridization
○ NanoString GeoMx
○ Nanostring과 10x Genomics의 특허 분쟁 ('23) (ref1, ref2) → Nanostring 파산 (ref) 및 인수 (ref)
○ Stereo-seq
○ flow cell 위에 새겨진 oligo patterning을 Illumina 또는 MGI sequencing으로 읽어들이고 barcode calling
○ 직경 : 220 nm
○ 스팟 간 거리 : 500 혹은 715 nm
○ Seq-Scope : Visium보다 해상도가 높음
○ flow cell 위에 새겨진 oligo patterning을 Illumina 또는 MGI sequencing으로 읽어들이고 barcode calling
○ PIC(photo-isolation chemistry)
○ PIXEL-seq
○ XYZeq (직경 500 μm)
○ spatially barcoded microwell 위에 조직을 올려놓기 → 1회 역전사 → 세포 제거 → single-cell sequencing
○ sci-Space (sc-space; 직경 222 μm)
○ spatially gridded hashing oligo가 있는 슬라이드 위에 조직을 올려놓기 → tissue permeabilization (oligo transfer) → 이미징 → 세포핵 제거 → 세포 고정 → 시퀀싱
○ sci-RNA-seq
○ TIVA-seq
○ NICHE-seq
○ ZipSeq
○ 세포에 photocaged oligonucleotides를 처리하여 실시간으로 RNA들의 patterned illumination (i.e., zipcode)를 관찰하는 이미징 기술
○ DBiT-seq
○ orthogonal microfluidics 상의 일정한 위치 위에 barcoded oligo를 올려서 조직의 위치 특이적 전사체를 획득
○ Slide-Tags (직경 10 μm)
○ CITE-seq (ref 1, ref 2) : 공간전사체와 antibody의 분포를 나란히 비교할 수 있음

Figure. 12. CITE-seq 모식도
○ streptavidin-biotin을 이용하여 oligonucleotide의 5' 말단을 항체에 연결
○ oligonucleotide는 oligo-dT 프라이머와 상보적으로 결합할 수 있음
○ streptavidin-biotin 결합은 환원 조건에서 해리될 수 있음
○ 최근 perturb-CITE-seq 기술도 개발됨
○ SPOTS
○ Spatial PrOtein and Transcriptome Sequencing
○ polyadenylated DNA-barcoded antibody를 이용하여 Visium 상에서 protein level을 간접적으로 확인
○ Open-ST
○ MAGIC-seq
⑤ 2-2. 이미지 기반 공간전사체 (image-based spatial transcriptomics) : 적은 유전자 + 많은 스팟
○ ISS(in situ sequencing) : 조직에 RNA가 있던 자리에서 RNA가 시퀀싱되도록 하는 기술. sequencing by ligation
○ 종류 1. 최초의 ISS
○ 종류 2. ISS with Padlock probe
○ reverse transcriptase는 RNA 타겟의 cDNA를 만듦
○ Padlock probe는 cDNA의 두 개 영역에 혼성화할 수 있음
○ 타겟 서열의 증폭은 RCA(rolling-circle amplification)을 통해 이루어짐
○ RCA product는 ligation에 의해 in situ에서 시퀀싱됨
○ 종류 3. ISS using fluorescent probes and cross-linking
○ 종류 4. barcode based methods
○ 종류 5. gap-filled ISS
○ FISH
○ smFISH(single molecule FISH) (2008년)
○ seqFISH(sequential FISH) (2014년) : DNAse I 처리와 연속적인 염색 및 이미징을 통해 각 RNA transcript 신호를 획득함. 12개의 pseudocolor를 사용하고 각 색상은 4 라운드가 필요함. 각 바코드는 5개의 pseudocolor를 포함
○ seqFISH+ : 각 encoding round 별 20 probe를 쓰고 형광 구간을 잘 나눠써서 게놈 스케일로 전사체를 얻는 기술
○ Vizgen - MERSCOPE (기술명 : MERFISH (multiplexed error-robust FISH)) : ISH 기반
○ direct probe hybridization + 별도의 ampliciation 메커니즘 없음
○ 각 FISH probe는 각 유전자와 1:1 대응 (단, 이 가정이 완전히 성립하지는 않을 수 있음)
○ barcode assignment(i.e., barcode calling)에 있어서 error correction 방법을 사용함 : 해밍 코드
○ 단계 1. FISH probe 별로 시간에 따라 형광 여부를 달리하여 여러 사진을 촬영
○ 단계 2. 각 RNA에서 읽혀진 이진 코드를 통해 역으로 어떤 유전자인지를 추측
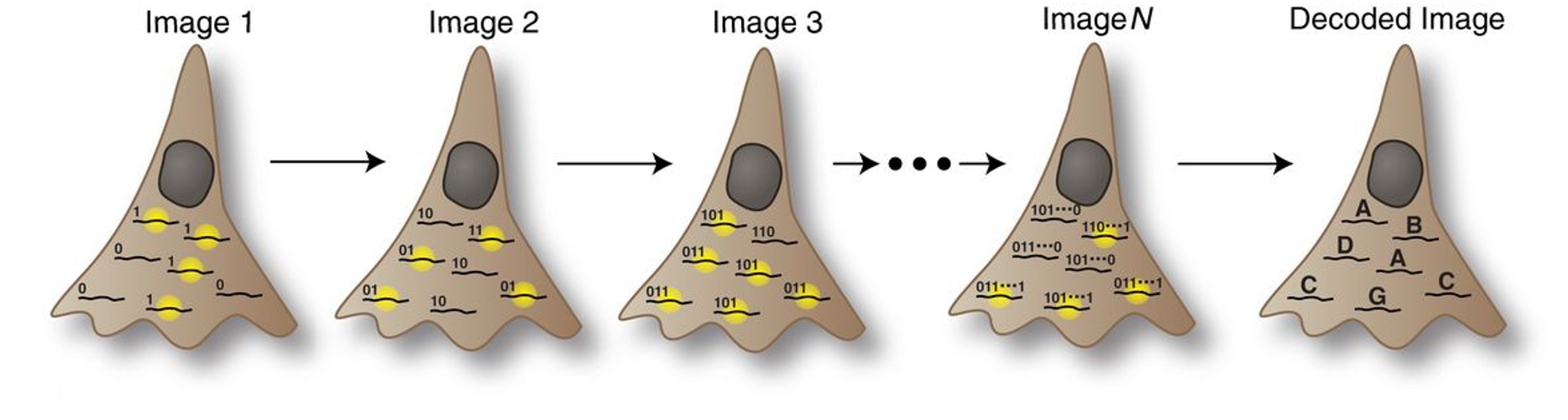
Figure. 13. MERFISH의 원리
○ 10x Genomics - Xenium : ISS 기반
○ 소량의 padlock probe + RCA(rolling circle amplification)
○ 단계 1. padlock probe가 상보적인 RNA transcript를 집게 모양으로 결합한 후 고리를 형성
○ 단계 2. RCA(rolling circle amplification) : 고리가 형성된 뒤 해당 RNA transcript가 증폭됨
○ 단계 3. 각 RNA transcript를 형광 probe로 혼성화시킨 뒤 형광 이미징 → washing
○ 단계 4. 단계 3을 반복한 뒤 생성된 이미지로부터 각 유전자에 대한 레이블로 decoding

Figure. 14. Xenium의 원리
○ Nanostring - CosMx : ISH 기반
○ 소량의 probe + branch chain hybridization
○ Nanostring과 10x Genomics의 특허 분쟁 ('23) (ref1, ref2) → Nanostring 파산 (ref) 및 인수 (ref)
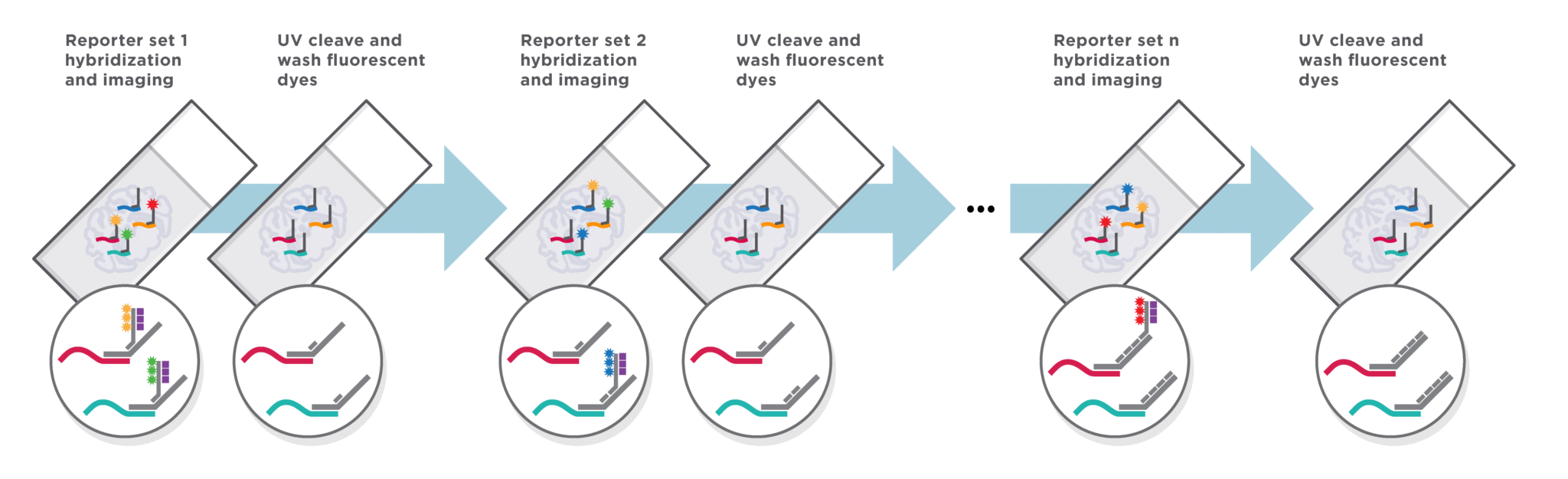
Figure. 15. CosMx의 원리
○ FISSEQ 및 oligoFISSEQ
○ Veranome
○ Rebus
○ BOLORAMIS
○ STARmap 및 STARmap PLUS : sequencing by ligation
○ SEDAL sequencing
○ ExSeq
○ BaristaSeq : sequencing by synthesis
○ BARSeq 및 BARSeq2
○ HybISS
○ SABER
○ clampFISH
○ split-FISH
○ SCRINSHOT
○ PLISH
○ osmFISH
○ ExFISH
○ par-seqFISH
○ EASI-FISH
○ SGA
○ corrFISH
○ EEL FISH
⑥ 종류 3. 공간 단백질체(spatial proteomics) : 크게 질량 분석법 기반과 이미징 기반으로 구분
○ SWITCH
○ MxIF
○ t-CyCIF
○ IBEX
○ DEI
○ CODEX
○ immuno-SABER
○ TSA
○ Opal IHC
○ MIBI
○ IMC
○ HD-MIBI
○ GeoMx Digital Spatial Profiler (DSP)
○ 100 mm 스케일, 10 um 해상도, ISH 기반
○ UV-cleavable DNA barcode와 결합된 항체 혹은 유전자 프로브를 이용
⒃ 기타 시퀀싱 기술
① TCR-seq (T cell receptor sequencing) : T cell 서브타입 및 클론을 추적하기 위한 시퀀싱
② Invade-seq : host-microbiome을 분석하기 위한 시퀀싱 기술
③ long-read sequencing : 2022년 올해의 기술 (레퍼런스)
| short-read seq | long-read seq | |
| 출시 년도 | 2000년대 초반 | 2010년대 중반 |
| 평균 리드 길이 | 150-300 bp | 5,000-10,000 bp |
| 정확도 | 99.9% | 95-99% |
Table. 3. short-read seq과 long-read seq의 비교
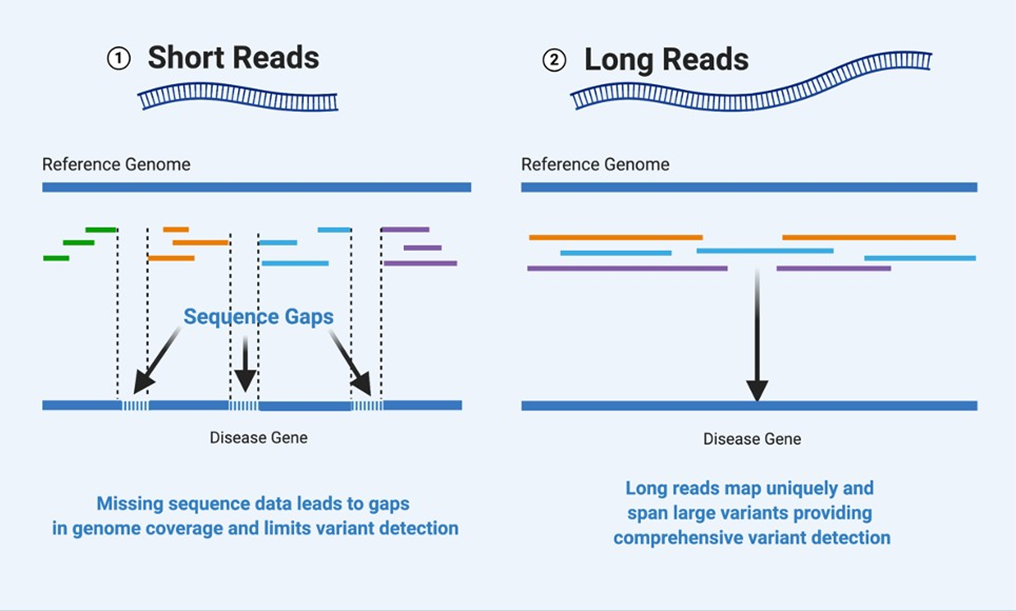
Figure. 16. short-read seq과 long-read seq의 차이
○ 시퀀싱 갭이 없어 다음과 같은 분석을 수행할 수 있음
○ 장점 1. AS 분석(alternative splicing analysis) : alternative splicing event, isoform 등에 대해서도 식별 가능해짐
○ 장점 2. CNV 분석(copy number variation analysis) : 예를 들어, 헌팅턴 무도병은 반복서열의 수가 중요함
○ 장점 3. epigenetics와 transcriptomics의 결합도 용이해짐
○ 종류 1. Pacific Biosciences SMRT(single molecule real-time) sequencing : 평균 read 길이는 ~20 kb
○ 종류 2. Oxford Nanopore Sequencing
○ 평균 리드 길이 : ~ 100 kb
○ 최대 리드 길이 > 2 Mb
○ 높은 에러율 : 5-10%. 참고로, Illumina sequencing의 에러율은 0.1-1% 정도
④ non-invasive sequencing
○ cell을 깨지 않고 시퀀싱 할 수 있는 기술
⑤ Halo-seq : 특정 타겟과 인접한 RNA들의 전사체를 얻는 기술
○ 1단계. 특정 타겟에 HaloTag domain을 붙임
○ 2단계. 해당 HaloTag는 radical-producing Halo ligand로서 주입한 alkyne handle로부터 수소 라디칼 H∙을 이탈시켜 alkyne handle 라디칼을 생성
○ R-H → R∙ + H∙
○ 3단계. 비슷하게 HaloTag는 RNA로부터 수소 라디칼 H∙을 이탈시켜 RNA 라디칼을 생성
○ RNA-H → RNA∙ + H∙
○ 4단계. alkyne handle 라디칼과 RNA 라디칼이 결합
○ 5단계. alkyne-RNA와 biotin azide를 반응시켜 biotinylated RNA를 생성
○ 6단계. streptavidin을 이용한 affinity chromatography로 biotinylated RNA만을 분리
○ 7단계. RNA-seq을 통해 특정 타겟과 가까운 RNA만을 탐지할 수 있음
○ 이유 : 라디칼은 불안정하므로 먼 거리를 이동하지 못함
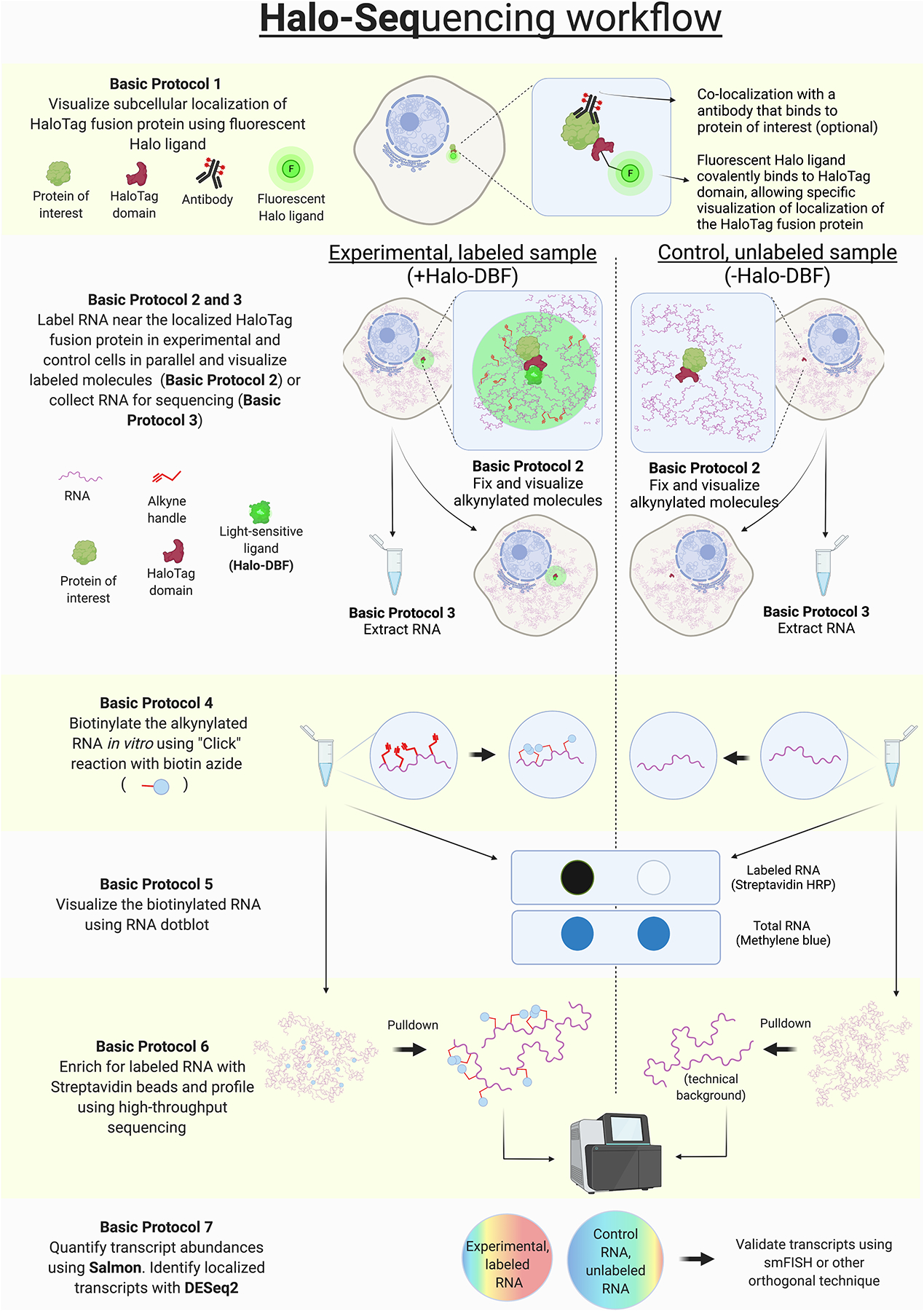
Figure. 17. Halo-seq의 원리
⑥ multi-NTT seq (nanobody tethered transposition followed by sequencing)
⑦ 후성유전체 시퀀싱(epigenomics sequencing)
⑧ 3차원 시퀀싱
⑨ temporal sequencing
○ Live-seq
○ TMI
⑩ 시공간 오믹스
○ ORBIT (single-molecule DNA origami rotation measurement)
○ 4D spatiotemporal MRI 또는 hyperpolarized MR
○ in vivo 4D omics with transparent mice
⒄ NGS(next-generation sequencing) 요약
① 유전체 분석의 비용
○ 2001년 : 인간 게놈 프로젝트 기준 $100 million / person
○ 2007년 : 1000억 원 / 4년
○ 2008년 : 454 Life Sciences 기준 $1,000,000 / person. 15억 원 / 4.5개월
○ 2009년 : Helicos BioSciences 기준 $48,000 / person
○ 2014년도에는 백만 원이면 충분할 것으로 예측 (Nature 456, 23-25, 2008)
② 유전체 분석의 규모
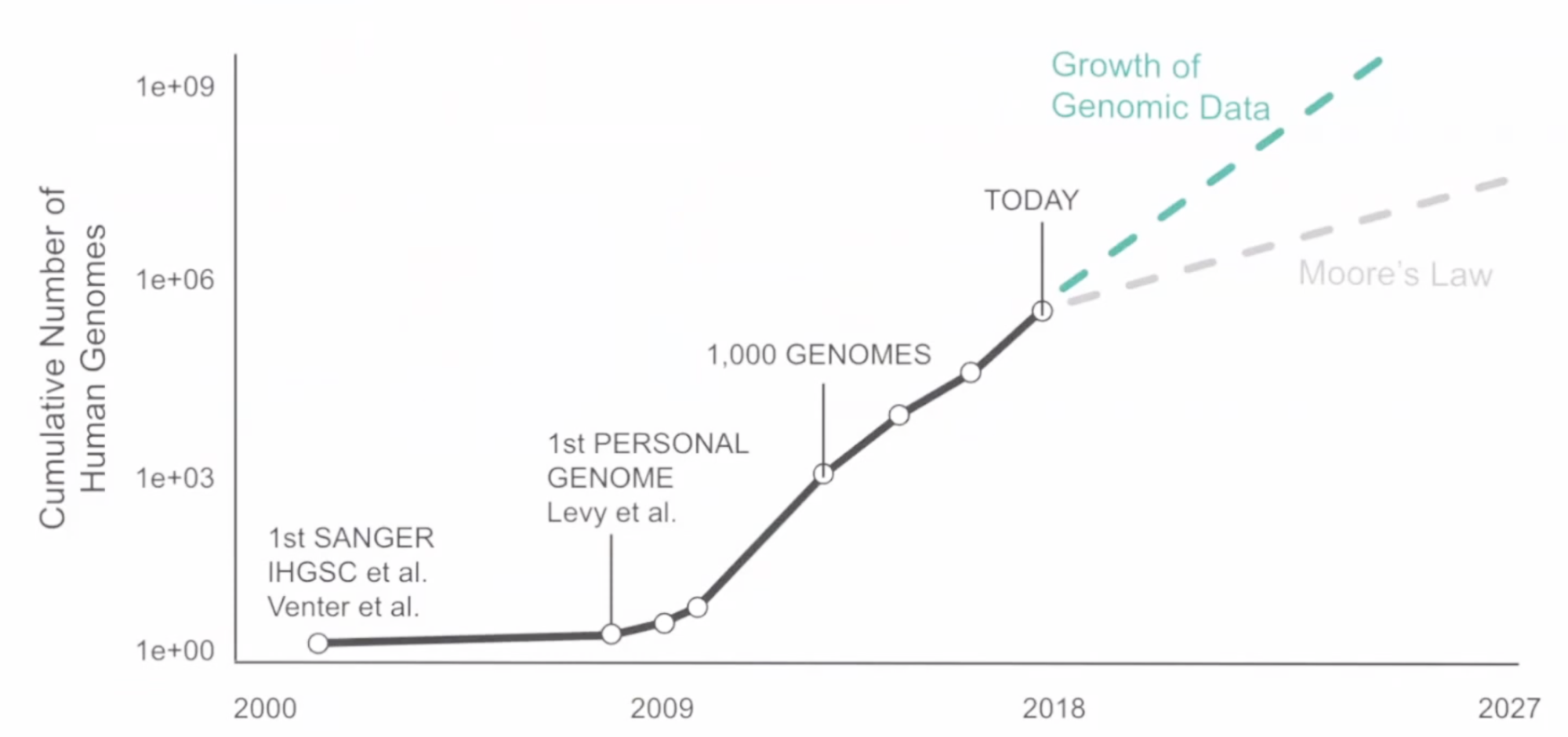
③ depth와 coverage의 관계
○ sequencing depth (read depth) : 특정 뉴클레오티드가 평균적으로 몇 번 나타나는지를 의미
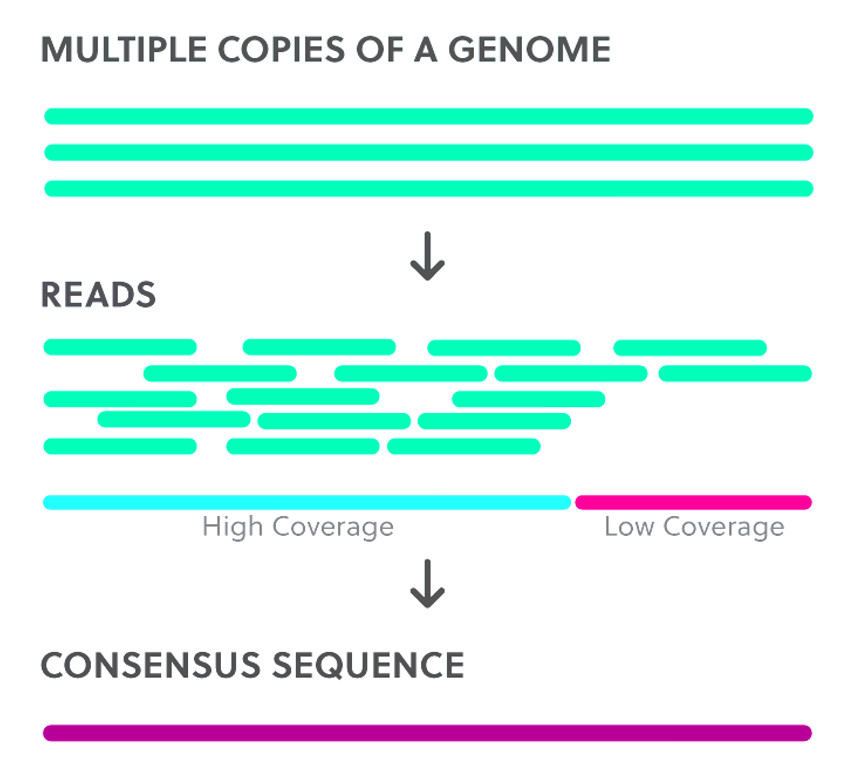
Figure. 19. depth의 정의
○ "10x"는 10번 반복하여 읽었음을 의미
○ 뉴클레오티드별로 정의될 수 있음
○ coverage (c)
○ c := LN / G
○ L : read length
○ N : number of reads
○ G : haploid genome length
○ total read number에 대해서는 sequencing depth로 표현
○ sequence reads와 reference (예 : whole genome, al locus)에 대한 관계에 대해서는 coverage로 표현
○ 그 이외에는 depth와 coverage는 굉장히 유사한 개념이라고 할 수 있음
④ bulk와 read의 관계
○ bulk : total RNA production
○ depth가 동일한 경우 bulk가 커짐에 따라 RNA read count가 반비례하는 불합리가 발생
○ 예 : spatial transcriptomics의 경우 대표적으로 bulk가 크고 depth가 낮아 RNA read count가 낮음
○ 정규화 : 위 불합리를 해소하기 위해 여러 방법이 도입됨
⑤ read count와 number of reads의 관계
○ read length가 250 bp 미만인 경우 sequence error를 탐지할 수 없음
○ read length와 number of reads per run의 관계 : trade-off가 있음
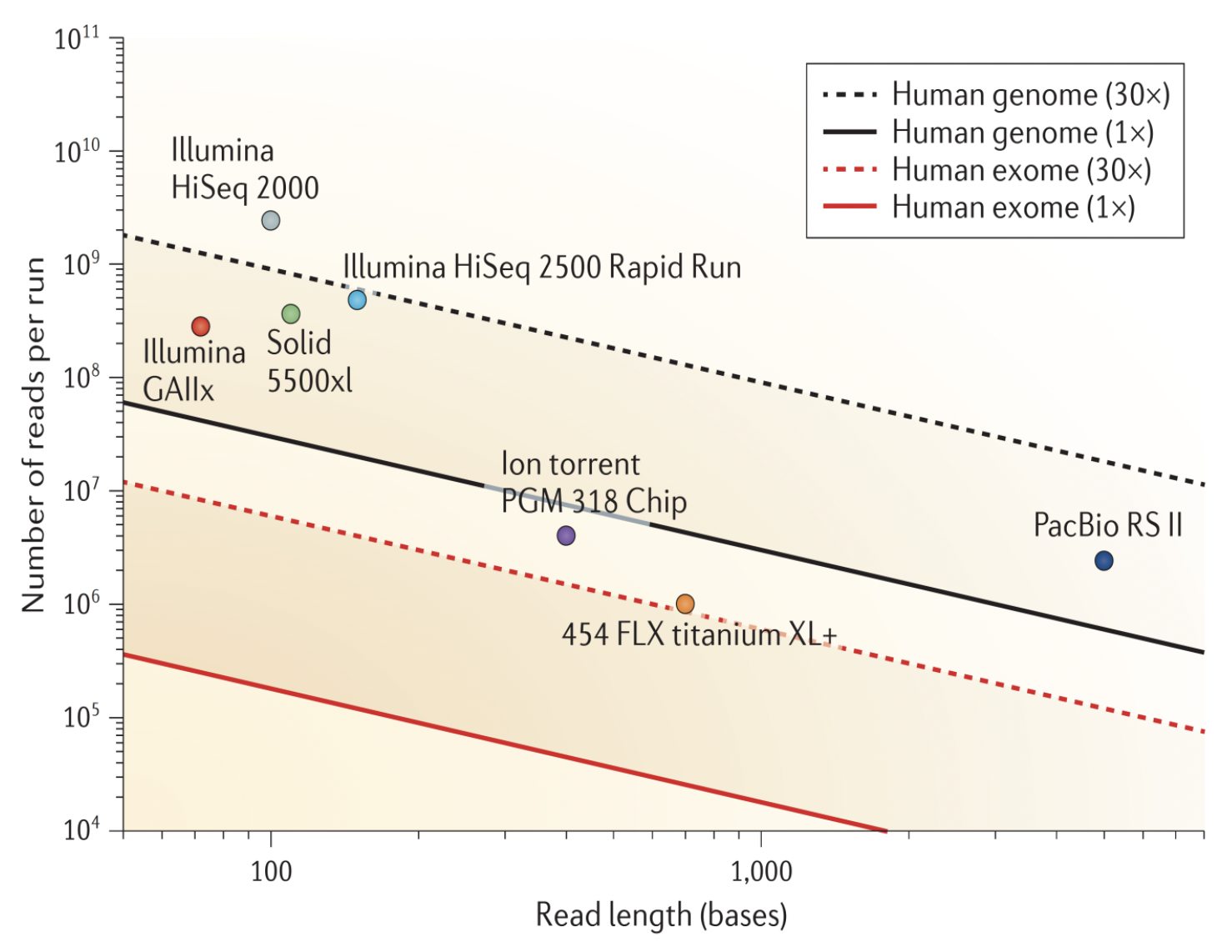
Figure. 20. read length와 number of reads per run의 관계
⑥ 전사체의 read count와 gene expression의 관계
○ read count : 실제 transcripts의 개수
○ gene expression : 정규화 과정(normalization)을 통해 read count에서 보정한 값
입력: 2015.07.02 23:31
수정: 2025.02.02 13:11
'▶ 자연과학 > ▷ 일반생물학' 카테고리의 다른 글
| 【생물학】 Siglec (1) | 2024.04.09 |
|---|---|
| 【생물학】 23강. 의학 (0) | 2023.06.04 |
| 【생물학】 8-2강. 미생물학의 중심학설 (0) | 2022.07.12 |
| 【생물학】 암세포 휴면 상태(cancer cell dormancy) (0) | 2022.06.18 |
| 【생물학】 6강. 신호전달 (0) | 2022.06.13 |



최근댓글